 While absolutely essential to modern medicine, broad-spectrum antibiotics use has contributed to the rise in antibiotic resistance and, via perturbations of the gut microbiota, adversely impacted some aspects of human health. To address these issues, there has been recent work on the development of precision antibiotic agents that only kill pathogenic bacteria.
While absolutely essential to modern medicine, broad-spectrum antibiotics use has contributed to the rise in antibiotic resistance and, via perturbations of the gut microbiota, adversely impacted some aspects of human health. To address these issues, there has been recent work on the development of precision antibiotic agents that only kill pathogenic bacteria.
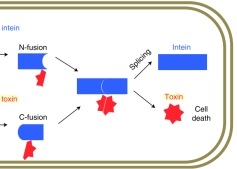
López-Igual et al. describe an engineered toxin-intein antimicrobial that is only activated in the presence of specific transcription factors unique to the targeted pathogen. In their paper, they demonstrate killing of an antibiotic-resistant strain of Vibrio cholerae using this method and note the potential for their approach to reduce the occurrence of antibiotic resistance.
At journal club we will be discussing:
a) briefly and broadly, the need for precision antibiotic agents and issues with various approaches to providing such therapy (e.g. novel small molecules, phages, etc.)
b) how do the authors target specific pathogens and how effective is it?
c) can we apply similar engineering approaches to modulate the microbiota in other ways?
The final human microbiome journal club of 2019 will be on December 6 at 3PM in MUMC4N55A. All are welcome!
Paper citation: López-Igual, R., Bernal-Bayard, J., Rodríguez-Patón, A., Ghigo, J. M. & Mazel, D. Engineered toxin–intein antimicrobials can selectively target and kill antibiotic-resistant bacteria in mixed populations. Nat. Biotechnol. (2019). doi:10.1038/s41587-019-0105-3